Our trained hearing care specialists can assist you with finding the ideal hearing aid style and customization options to meet your individual needs. We offer some of the best brands on the market, including Phonak, Signia, Oticon, and ReSound.
Our mission is to provide all individuals with market-leading hearing aid technology so that they can enjoy the quality of life that they deserve.


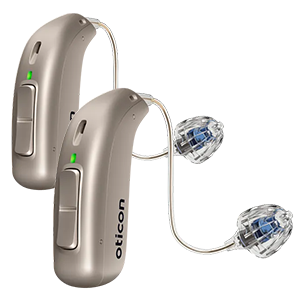


When it comes to customization, Phonak is all about it. Offering a large variety of hearing aid models and styles, Phonak users can select a custom hearing aid that matches their needs. One of the nicest features that Phonak currently offers is an invisible, in-the-ear hearing aid that performs for months before needing to be properly recharged.
One of the most common brands you'll hear about is Signia, co-branded with Siemens. These devices offer top-quality sound and noise control in any environment, thanks to their unique Primax technology. Signia offers a RIC fit and has a whole range of customization options so that you can get a device that fits your needs and wants.
Oticon is a worldwide hearing aid brand that has been around for a long time. Over the years, they've continued to include more technology in their hearing devices. Some of the most desired are Bluetooth streaming, OpenSound Navigator, and speech rescue. Oticon provides many different styles, features, and designs in their lineup, including IIC, ITC, ITE, RITE, and BTE.

Today there are digital hearing aids to complete your convenient lifestyle. When technology works intuitively, you barely notice it is there. ReSound has been a pioneer in building innovative hearing solutions and makes it easier and more comfortable than before to access personalized sound that suits your lifestyle. Hearing aid technology has come a long way from bulky devices of the past.

Our current product lineup offers both behind-the-ear and receiver-in-ear hearing aids. Whether you're looking for a basic option or a discreet one, we can match you with the right hearing aid to fit your needs. We know that all customers don't have the same conditions. For this reason, it's vital to have variety so that everyone can choose the customization options that meet their individual needs.
Apart from hearing aids, we also offer custom hearing care products. Some of the most widely desired are in-ear monitors, swimming plugs, custom impressions, shooter ears, and ear protection. Simply stop by one of our offices, and we'll be more than happy to assist you with any of these custom hearing care products.




The FD&C Act establishes a comprehensive system for the regulation of devices, as defined in section 201(h) of the FD&C Act (21 U.S.C. 321(h)), intended for human use. Section 513 of the FD&C Act (21 U.S.C. 360c) defines three classes of devices, reflecting the regulatory controls needed to provide reasonable assurance of their safety and effectiveness. The three classes of devices are class I (general controls), class II (special controls), and class III (premarket approval) (see 21 U.S.C. 360c). Hearing aids are devices intended for human use and are subject to the FD&C Act. Currently, air-conduction hearing aids are generally either class I or class II devices.
The FD&C Act also directs the establishment of an electronic product radiation control program under section 532(a) to protect the public health and safety (see 21 U.S.C. 360ii(a)), and requires, among other things, that manufacturers of electronic products provide notification of certain defects (see 21 U.S.C. 360 ll). Section 531(1)(B) of the FD&C Act defines electronic product radiation as, among other phenomena, any sonic, infrasonic, or ultrasonic wave emitted from an electronic product as the result of the operation of an electronic circuit (see 21 U.S.C. 360hh(1)(B)). In turn, any manufactured or assembled product which, when in operation, contains or acts as part of an electronic circuit and emits (or in the absence of effective shielding or other controls would emit) electronic product radiation would be an electronic product (see 21 U.S.C. 360hh(2)(A)). As such, hearing aids and PSAPs emit electronic product radiation and are electronic products, meaning they are subject to the electronic product radiation control requirements.
FDARA amended the FD&C Act to apply requirements specific to certain hearing aids and defined the term “over-the-counter hearing aid” (see 21 U.S.C. 360j(q)). We are issuing these requirements for OTC hearing aids pursuant to section 709(b) of FDARA, which authorizes FDA to establish requirements for labeling, output limits, conditions for sale and distribution of OTC hearing aids, and other requirements that provide for reasonable assurance of safety and effectiveness of these devices.
In addition, the FD&C Act provides that a device is misbranded unless, among other requirements, its labeling bears adequate directions for use (see section 502(f)(1) of the FD&C Act (21 U.S.C. 352(f)(1)). Consistent with section 502 of the FD&C Act, FDA has issued regulations that exempt certain kinds of devices from the requirement for adequate directions for use. Section 502(f)(2) further requires adequate warnings against use of a device in those pathological conditions, or by children, where use of the device may be dangerous to health. The labeling must also bear adequate warnings against unsafe dosage or methods or duration of administration or application (see section 502(f)(2) of the FD&C Act). Such warnings must be in such manner and form as are necessary for the protection of the users (see section 502(f)(2) of the FD&C Act).
A device is also misbranded if its labeling is false or misleading in any particular (see section 502(a) of the FD&C Act). Section 201(n) of the FD&C Act states that in determining whether labeling or advertising is misleading, there shall be taken into account not only representations made or suggested but also the extent to which labeling or advertising fails to reveal material facts.
Other misbranding provisions under the FD&C Act would apply as well, including section 502(c), which deems a device to be misbranded if any word, statement, or other information required by or under authority of the FD&C Act to appear on the label or labeling is not prominently placed thereon with such conspicuousness and in such terms as to render it likely to be read and understood by the ordinary individual under customary conditions of purchase and use.
Additionally, section 701(a) of the FD&C Act authorizes FDA to issue regulations for the efficient enforcement of the FD&C Act (21 U.S.C. 371(a)). The regulations established in this rulemaking are for the efficient enforcement of the FD&C Act because they will provide standards for the legal marketing of safe and effective hearing aids.
Violations of any final rules from this rulemaking, once in effect, would render the hearing aids adulterated and/or misbranded under sections 501 and/or 502 of the FD&C Act, and subject to enforcement action, for example, seizure (see section 304 of the FD&C Act (21 U.S.C. 334)), injunction (see section 302 of the FD&C Act (21 U.S.C. 332)), and criminal prosecution (see section 303 of the FD&C Act (21 U.S.C. 333)). Prohibited acts include, among others, introducing an adulterated or misbranded device into interstate commerce (see section 301 of the FD&C Act (21 U.S.C. 331)). Sections 538 and 539 of the FD&C Act additionally set forth prohibited acts and provisions for enforcement for electronic product radiation control (see 21 U.S.C. 360 oo and 360pp, respectively).
Under section 521 of the FD&C Act, no State or political subdivision of a State may establish or continue in effect with respect to a device intended for human use any requirement that is different from, or in addition to, any requirement applicable under the FD&C Act to the device and that relates to the safety or effectiveness of the device or to any other matter included in a requirement applicable to the device under the FD&C Act (21 U.S.C. 360k). Section 521 of the FD&C Act also provides that FDA may grant an exemption from preemption under certain circumstances. Section 709(b) of FDARA also includes a preemption provision with respect to requirements for OTC hearing aids.
A device which, because of any potentiality for harmful effect, or the method of its use, or the collateral measures necessary to its use is not safe except under the supervision of a practitioner licensed by law to direct the use of such device, and hence for which “adequate directions for use” cannot be prepared, shall be exempt from section 502(f)
(1) of the act if all the following conditions are met:
(a) The device is:
(1)
(i) In the possession of a person, or his agents or employees, regularly and lawfully engaged in the manufacture, transportation, storage, or wholesale or retail distribution of such device; or
(ii) In the possession of a practitioner, such as physicians, dentists, and veterinarians, licensed by law to use or order the use of such device; and
(2) Is to be sold only to or on the prescription or other order of such practitioner for use in the course of his professional practice.
(b) The label of the device, other than surgical instruments, bears:
(1) The symbol statement “Rx only” or “℞ only” or the statement “Caution: Federal law restricts this device to sale by or on the order of a ___”, the blank to be filled with the word “physician”, “dentist”, “veterinarian”, or with the descriptive designation of any other practitioner licensed by the law of the State in which the practitioner practices to use or order the use of the device; and
(2) The method of its application or use.
(c) Labeling on or within the package from which the device is to be dispensed bears information for use, including indications, effects, routes, methods, and frequency and duration of administration, and any relevant hazards, contraindications, side effects, and precautions under which practitioners licensed by law to administer the device can use the device safely and for the purpose for which it is intended, including all purposes for which it is advertised or represented: Provided, however, That such information may be omitted from the dispensing package if, but only if, the article is a device for which directions, hazards, warnings, and other information are commonly known to practitioners licensed by law to use the device. Upon written request, stating reasonable grounds therefor, the Commissioner will offer an opinion on a proposal to omit such information from the dispensing package under this proviso.
(d) Any labeling, as defined in section 201(m) of the act, whether or not it is on or within a package from which the device is to be dispensed, distributed by or on behalf of the manufacturer, packer, or distributor of the device, that furnishes or purports to furnish information for use of the device contains adequate information for such use, including indications, effects, routes, methods, and frequency and duration of administration and any relevant hazards, contraindications, side effects, and precautions, under which practitioners licensed by law to employ the device can use the device safely and for the purposes for which it is intended, including all purposes for which it is advertised or represented. This information will not be required on so-called reminder - piece labeling which calls attention to the name of the device but does not include indications or other use information.
(e) All labeling, except labels and cartons, bearing information for use of the device also bears the date of the issuance or the date of the latest revision of such labeling.
[41 FR 6896, Feb. 13, 1976, as amended at 81 FR 38930, June 15, 2016]